Authors: Jeremy D. Parsons and Patricia Rodriguez-Tomé
EMBL-Outstation - The European Bioinformatics Institute (EBI), Wellcome
Trust Genome Campus, Hinxton, Cambridge CB10 1SD, UK;
Corresponding Author:
Jeremy D. Parsons
Boinformatics Consultancy
37 Walpole Road,
Cambridge,
CB1 3TH
England
Tel. +44 7986 534110
http://www.littlest.co.uk/
JESAM: CORBA software components to create and publish EST alignments and
clusters
Abstract
Motivation:
Expressed Sequence Tags (ESTs) are cheap, easy and quick to obtain relative
to full genomic sequencing and currently sample more eukaryotic genes than
any other data source. ESTs are particularly useful for developing Sequence
Tag Sites (STSs for mapping), polymorphism discovery, disease gene hunting,
mass spectrometer proteomics, and most ironically for finding genes and
predicting gene structure after the great effort of genomic sequencing.
However, ESTs have many problems and the public EST databases contain all
the errors and high redundancy intrinsic to the submitted data so it is
often found that derived database views, which reduce both errors and redundancy,
are more effective starting points for research than the original raw submissions.
Existing derived views such as EST cluster databases and consensus databases
have never published supporting evidence or intermediary results leading
to difficulties trusting, correcting, and customizing the final published
database. These difficulties have lead many groups to wastefully repeat
the complex intermediary work of others in order to offer slightly different
final views. A better approach might be to discover the most expensive
common calculations used by all the approaches and then publish all intermediary
results. Given a globally accessible database with a suitable component
interface, like the JESAM software described in this paper, the creation
of customized EST-derived databases could be achieved with a minimum effort.
Results:
Databases of EST and full-length mRNA sequences for four model organisms
have been self-compared by searching for overlaps consistent with contiguity.
The sequence comparisons are performed in parallel using a PVM process
farm and previous results are stored to allow incremental updates with
minimal effort. The overlap databases have been published with CORBA interfaces
to enable flexible global access as demonstrated by example Java applet
browsers. Simple cDNA supercluster databases built as alignment database
clients are themselves published via CORBA interfaces browsable with prototypical
applets. A comparison with UniGene Mouse and Rat databases revealed undesirable
features in both and the advantages of contrasting perspectives on complex
data.
Availability:
The software is packaged as two jar files available from: URL: http://www.littlest.co.uk/
. One jar contains all the Java source code, and the other contains all
the C, C++ and IDL code. Links to working examples of the alignment and
cluster viewers (if remote firewall permits) are no longer available at http://www.ebi.ac.uk/EST.
Contact:
http://www.littlest.co.uk/
Introduction
CORBA and biological data
The use of CORBA in a biological context was introduced by Hu et al. (1998)
and Lijnzaad et al. (1998) where they explain that CORBA can be a good
solution to the problem of creating applications for distributed heterogeneous
environments. The Internet is the extreme example of both distribution
and heterogeneity and, therefore, the hardest to support with conventional
client/server systems. Orfali and Harkey (1998) see CORBA as a good
middleware solution for the Internet and refer to the creation of the "Object
Web" where CORBA hides some of the most complex interoperability issues
behind well specified interfaces fronting globally accessible software
components. Principal among the benefits that CORBA brings to biological
data distribution and interaction, are: the Interface Definition Language
(IDL) to define interfaces between objects, scalability via language, location
and Operating System independence, and, finally, a rich set of 15 object
services. CORBA is often described as being middleware because it sits
between software components, or data sources, and lets them all interact
without regard to intervening distance or data conversions. In 1996, the
ease with which client/server object-oriented applications could be written,
distributed, and supported across the Internet increased when Netscape
(http://www.netscape.com/) announced (Orfali and Harkey, 1997) that its
browsers were all going to include a CORBA Object Request Broker (ORB)
alongside the built-in Java Virtual Machine (JVM), and when it subsequently
decided to distribute its browsers for free.
Sequence databases (for example the EMBL NSDB Stoesser et al, 1998)
are currently the most familiar biological database and a typical source
for EST sequences. Traditionally these databases are presented and distributed
using "flat file" views of the sequence originally submitted, but these
static raw views are being superseded by novel interfaces better suited
to the needs of academic and industrial researchers. The new browsing interfaces
are needed because current research may need to investigate nucleotide
or protein sequences grouped by their function, phylogeny, map position,
cellular location, expression regulation, disease association, annotation
similarity or metabolic pathway etc. To support these new investigative
cross-database clients, it is useful to have standardized API level access
to both data and services - a requirement where CORBA is starkly appropriate.
In summary, ESTs are inherently difficult data to handle (yet extremely
useful) so they are a natural target for new efforts to improve data quality,
cross database browsing and novel visualization tools.
EST databases and clustering extensions
Of all the biological databases, Expressed Sequence Tag (EST)
databases have been the most conspicuous in recent years both because of
their prodigious growth rates and because of their increase in intrinsic
importance connecting different areas of biological research. Expressed
Sequence Tags are cheap, easy and quick to obtain relative to full genomic
sequencing and currently sample more eukaryotic genes than any other data
source. They have great current utility for disease gene hunting (Banfi
et al. 1996), mass spectrometer proteomics (Kuster and Mann, 1998), and
for finding genes and predicting gene structure after genomic sequencing
(Bailey et al, 1998, and Jiang and Jacob, 1998). The patterns of early
public work with human ESTs (Boguski et al, 1993; Hillier et al, 1996;
Gerhold and Caskey, 1996) are being repeated again now for mouse and other
eukaryotic model species. The Mouse Genome Project is advancing quickly
(Bettey et al, 1999), with a large EST project (Marra et al, 1999) and
various maps (Blake et al, 1998, McCarthy et al, 1997) completed and novel
databases available for unique analyses of mammalian development
(Davidson et al, 1997), and inter-species comparisons (Hardison, et al,
1997, Makalowski et al, 1996).
ESTs have many problems that stem from their means of production. The
cDNA library from which any ESTs are drawn will sample the levels of expression
in a particular tissue at a particular time: rare transcripts will be missed
and highly expressed genes will be overly abundant. This latter problem,
redundancy, is both wasteful and difficult to handle due to the extreme
volume of error prone data. Redundancy can be reduced in the laboratory
using normalization techniques but these are imperfect though additional
subtractive techniques are being investigated (Bonaldo et al, 1996).
To address the redundancy problem using computer techniques, a variety
of EST clustering programs have been implemented for example Parsons et
al (1992), Boguski and Schuler (1995), Parsons (1995), Eckman et al. (1998),
and Yee and Conklin (1998). Once clustered, gene fragments can be assembled
using sequence assembly programs, or both steps can be combined in large-scale
EST assembly and gene indexing protocols. An assembly stage adds two main
benefits: first, it produces contigs and consensus sequences which can
completely hide EST redundancy and, second, it should also improve the
length and quality of the gene reconstructions beyond that available from
any one EST (usually single pass reads). For some applications, the clustering
is all that is required, for example, the early stages of STS mapping but
for others, sophisticated cluster partitioning (Burke et al, 1998) or complex
assemblies are more suitable.
Whilst the redundancy problem can be mitigated, other deficiencies of
EST databases are computationally insoluble and most EST based projects
have centered on pragmatic scientific goals cognizant that perfect results
are impossible. Early projects that used EST clustering include TIGR's
THC database (Adams et al, 1995), Geneexpress (Houlgatte et al, 1995),
and UniGene (Boguski and Schuler, 1995) but new genomes (e.g. Arabidopsis,
Rounsley et al., 1996), new methods (e.g. SANBI STACK, http://www.sanbi.ac.za/Dbases.html),
and new public data (Aaronson, 1996) have kept the field busy with important
discoveries and enhanced views (Eckman et al, 1998).
Assembly and Gene Indexing
Assembling gene transcript fragments is more complex than assembling shotgun
clones of genomic DNA. One gene does not lead to one EST nor even a statistically
predictable sampling depth and, complicating further, there are also variations
in the underlying reconstruction target. The cDNA libraries that have been
used in the public EST projects were made from many different individuals/genotypes
and tissues so the public EST collections show polymorphism and tissue-specific
variation. Additionally, errors of many forms (basecalling, misnaming,
vector and cross-species contamination, chimeras, poor template, etc.)
are more likely with broadly sampled complex cDNA collections than with
tightly focussed genomic projects which can operate more internal checks
and aim for a simpler single target. Tissue variation may take the form
of alternative polyadenylation (Gautheret et al, 1998), or alternative
splicing (Lopez, 1998). In all cases, the best possible EST comparisons
will require knowledge of the likelihood of the various types of error,
and the exact tissue-specific origins of the source cDNA library.
EST clustering on typical cDNA/EST collections will produce clusters
which may vary in size from one to approximately 20,000 which is a size
range commonly encountered with larger shotgun assembly projects. Some
DNA shotgun assembly programs (necessary for all large-scale genomic sequencing)
have been applied to EST assembly, however there is a significant danger
of producing artificial sequences by incorrectly joining, or homogenizing
transcripts from different genes. In summary, postulated transcription
products will ultimately need experimental biological confirmation and
the totality of products from a single gene may not fit well into a FASTA
format file: instead, a description is needed with the potential to record
all of the reconstructed biological complexity.
EST clusters can be combined with other sources of gene sequence information,
(for example full-length mRNA, and genomic DNA) to better sample an organisms
entire genome, (especially for rare transcripts) to create "gene indexes",
and consensus sequence databases. As gene mapping information accumulates,
so the gene indexes lead to the creation of "gene maps" which link genes
and their functions to locations on a physical map. Gene maps supported
by polymorphism information make an extremely valuable resource for positional
candidate disease gene hunting.
Introduction to EST Clustering Algorithms
Computational Complexity
A typical EST sequencing project may produce between fifty thousand and
a few million ESTs which need to be compared against each other (order
N2). This formidable scaling problem must
be handled by whichever algorithm is chosen and before user considerations
such as input requirements and presentation style can be considered. Algorithmic
tools with the potential to reduce the comparison complexity include suffix
trees, hashes, Finite State Machines, indexing and statistical DNA-word
frequency analyses. These methods could be applied sequentially for each
sequence versus the rest, or (for some of the algorithms) a simultaneous
all-against-all result emerges inside some data structure.
Experimental and Biological Complexity
Deciding if and how two ESTs are related is complicated by errors in the
process of EST manufacture: short single pass reads (Hillier et al. 1996)
and by many natural causes of variation (Wolfsberg and Landsman, 1997).
The program that best estimates the likelihood of combinations of all these
complicating events should generate the best set of clusters or contigs,
and the best overall result. Given the scale and difficulty of the challenges
faced, even the best programs will make many mistakes and be unable to
assess the significance of, or just plain miss, much of importance so experimental
confirmation is necessary. This should not be a surprise as the same is
true of even fully finished genomes (Oliver, 1996).
Comparisons of Existing Programs
Unfortunately, there are as yet no detailed comparisons of the quality
of any current clustered EST databases and direct comparisons of the clustering
tools themselves are complicated by potentially important issues
such as results presentation style, software portability, memory, disk
and CPU requirements. Miller and Powell, (1994) published a quantitative
comparison of genomic shotgun assembly packages but this work is
not directly relevant because all the programs have since changed and the
paper did not address issues specific to EST assembly. Burke et al.(1998)
did publish two clustering comparisons: first a subpartitioning of UniGene101
but this used the existing (not independent) UniGene clusters as its starting
point and, second, a contrasting comparison with TIGR Gene Index but this
was summary and non-rigorous using different EST data.
Published references for EST comparison programs show a variety of specified
goals and assumptions. The simplest use scripting languages (e.g. Perl)
to run and parse sequence database searches by BLASTN (Altschul et al,
1997, http://www3.ncbi.nlm.nih.gov/BLAST/)
or FASTA (Pearson et al., 1997, http://www.cs.virginia.edu/brochure/profs/pearson.html).
EST sequences may be clipped to various levels of quality, vector trimmed,
and low complexity masked before being sent for BLASTN/FASTA comparisons.
BLASTN and FASTA hash the query sequence before scanning a sequence database.
JESAM and ACEmbly hash the whole database (indexing) and find hash hits
for the query against all database sequences simultaneously before checking
for alignments. Construction of Compugen's EST clusters (LEADS,
http://www.cgen.com/solutions/index.html)
uses a proprietary non-alignment based algorithm. Other (published) non-alignment
algorithms include EMBLSCAN (Bishop and Thompson, 1984) D-squared Cluster,
and RAPID (Miller et al, 1999) try to estimate the significance of discovering
oligos (DNA words) in common between two sequences without the cost of
a full alignment.
The simple ideal that all cognate ESTs should cluster together could
be achieved easily if all 3-prime ESTs were anchored to the same region
of the cDNA and gene. In this situation (ignoring sequencing errors) any
EST from the same gene would have the same basecalled sequence . The original
ICAtools (Parsons et al., 1992) were the first tools built to automate
this simple analysis. Unfortunately, as described by Gautheret et al. (1998),
alternate polyadenylation is common (in humans) so ESTs may not be well
anchored and discovering cognate ESTs reverts to solving a peculiar massive
shotgun assembly. The anchored EST assumption will, however, still give
useful results for strictly relative comparisons of the complexity within
individual cDNA libraries, especially if transcript heterogeneity is temporally
and anatomically restricted (Gautheret et al, Burke et al).
BLAST and FASTA based scripts
Examples of the BLASTN or FASTA clusterers include: INCA (Graul and Sadee,
1997, http://itsa.ucsf.edu/~gram/home/inca/)
SEALS (http://www.ncbi.nlm.nih.gov/Walker/SEALS/index.html),
WHALE (unpublished), Zymogenetics' REX (Yee and Conklin, 1998) and the
anonymous tools used to produce Geneexpress, and the Merck Gene Index.
The BLASTN/FASTA algorithms were designed for searches for local similarity
and homology, which is a different goal from a search for alignments consistent
with overlap/contiguity. In consequence, the script wrappers put around
the homology searches have three functions: to run the search, to parse
the search results and to enforce user-defined rules of when two sequences
should actually be considered to overlap. These extra match criteria cannot
be handled directly by the database searching programs so later, more sensitive
and slower pairwise sequence alignments may be run to improve the comparison
accuracy (Merck Gene Index). Typical extra heuristics include (for Geneexpress)
90% overall nucleotide identity in the overlapping region, no more than
35 unmatched (outside local alignment) dangling sequence ends (UniGene)
etc.
Glaxo's "Dynamic" assembler (Gill et al, 1997) uses interactive rounds
of BLASTN searches of EST databases to identify possible overlapping
sequences which are fed to Gap (Bonfield et al, 1995) for Greedy assembly.
The approach taken by Glaxo was quick to implement, and effective for simple
assemblies but suffers pauses between each round of assembly, the Greedy
assembly is prone to errors with repetitive sequence regions, the static
HTML output limits interactivity/editing and alternative BLASTN alignments
displayed simultaneously in one alignment may cause confusion for the user.
The original UniGene package (Boguski and Schuler, 1995, Schuler et
al, 1996) relied on a BLASTN-like hash based search to identify pairs of
sequences with at least two 13-mer words separated by no more than two
bases. These sequence pairs were then aligned more accurately within a
10-base window either side of the diagonal identified by the oligo match.
The alignment was scored +1 for a nucleotide identity, -2 for a mis-match,
-1 for a gap, and 0 for an N ambiguous base. An alignment quality was calculated
by dividing the score by the alignment length and had to exceed 0.91 to
be accepted. An extra constraint was that the aligned region had to extend
within 35 bases of the end of either sequence. UniGene itself only accepts
data of known good quality, (high quality base count, presence of poly-A
tail etc.) so not all ESTs that might pass the above alignment criteria
are ever checked for matches. Explicit lists of excluded sequences (>200,000)
are not published nor assemblies, alignments or consensus sequences. UniGene
is available online at http://www.ncbi.nlm.nih.gov/UniGene/index.html
with extra descriptions at http://www.ncbi.nlm.nih.gov/Schuler/Papers/ESTtransmap/
Specialized Clustering Programs
The ICAtools and CLEANUP are specialized hashed-query sequence clustering
packages. ICAass (one of the latest ICAtools - http://www.ebi.ac.uk/~jparsons)
uses a FASTA like search but uses an asymmetric scoring scheme designed
to measure redundancy, rather than directly discover overlaps. A variety
of specialized cluster browsing tools assist extraction of non-redundant
sequence sets, or allow accelerated database searches where an increased
portion of the computation time is spent comparing the query with very
similar sequences. All code is ANSI C, runs on many UNIX variants (and
ported to MacOS), and is free to academics and industry. It has been used
to cluster 180,000 ESTs on one shared 143 MHz UltraSPARC in 9 days. Memory
usage scales linearly with database size, but computation time scales quadratically.
CLEANUP (Grillo et al, 1996) is similar to the ICAtools but performs
a unique hashed-query pairwise sequence comparison which makes it slower
than ICAass. Query sequences are encoded as hashed oligos along with one
base mutated versions of the oligos to enhance query sensitivity. The largest
published data set: 2400 Drosophila sequences, was self-compared in 160
seconds. All code is written in C, and publicly available.
Small-Scale Assembly Programs
Conventional shotgun sequence assembly relies on overlapping alignment
of the called nucleotide bases to recreate a representation of the original
DNA progenitor sequence. All the shotgun assemblers referenced except ACEmbly
use a hashed query sequence algorithm for finding matches. Some of the
more sophisticated programs are able to make use of ancillary information
such as basecall probability estimates, clone orientation and length estimates
to improve the quality of the contigs produced, especially in the presence
of repetitive DNA. Relevant programs include Gap4, (Bonfield et al, 1995),
CAP3 (http://genome.cs.mtu.edu/cap/cap3.html),
Phrap and Gene Meyer's FAK (Meyers, 1995). The conventional shotgun assemblers
can be used to assemble typically-sized clusters of ESTs.
Gap4 ( http://www.mrc-lmb.cam.ac.uk/pubseq/overview.html
) implements a simple algorithm (relatively slow) where new shotgun reads
are compared against contig consensus sequences looking for hash hits that
extend the contigs in a Greedy fashion. The program is memory efficient
and scales to megabase-sized YAC assembly projects with more than 10,000
reads. Gap4 can read and extend contigs produced by other programs e.g..
Phrap, and CAP2.
Phrap (unpublished, http://bozeman.genome.washington.edu/index.html)
has incorporated error probability information deeper into assembly and
editing than any other assembly package. Alignments are stored in detail,
as a directed graph in RAM, leading to large memory requirements (1 GB
for a YAC assembly). A consensus sequence is extracted by using a mosaic
approach where the best read in any multiple alignment is directly copied
to the result. Phrap has no specific features to handle EST idiosyncrasies
like alternative splicing etc. Assemblies are fast (6 hours for 49,000
sequences on a single 195 MHz R10000) however a single sequence addition
requires all previous calculations to be repeated. An optimized and parallelised
version of Phrap is commercially available (http://www.spsoft.com/).
CAP3 (unpublished, http://genome.cs.mtu.edu/cap/cap3.html)
is an enhancement of CAP2 (Huang, 1996) - both are relatively slow. CAP3
can utilize ancillary information to guide sequence assembly: mate-pairs,
clone size, orientation/location, and basecall error estimates. CAP3 is
used at MIPS to assemble UniGene clusters (http://www.mips.biochem.mpg.de/proj/human/)
TIGR Assembler, (Sutton et al, 1995) is a relatively fast sequence assembly
program that has been proved assembling a diverse collection of repetitive
microbial genomes. Memory usage for the query hashing stage is 32 * 4n
bytes where n is the word size (oligo size) e.g. 512 MB for 12-mer oligos.
Four criteria control the match decision: minimum length of overlap, maximum
number of local errors, percentage of best possible score, and maximum
length of overhang. TIGR use a combination of BLASTN and TIGR Assembler
to produce their own consensus sequences. This combination is relatively
slow: multiple-months to produce THC (Tentative Human Consensus) sequences
for all the human ESTs. The THC database is biased towards splitting EST
clusters due to this stringent assembly stage (Burke et al.,1998). TIGR
Assembler on its own has been used to assemble 186,000 ESTs on a single
SPARC processor, with 512 MB RAM, in seven days.
ACEmbly (http://alpha.crbm.cnrs-mop.fr/acembly/)
is a fast hashed-database program where oligos are sampled from all sequences
and used to prime alignments and then assemblies. ACEmbly fragment ordering
is initially based on a distance metric calculated from the number of oligos
two sequences have in common divided by their total number of sampled oligos.
This "zipping" is fast (< 10 seconds for 600 reads) but prone to errors
with repeated sequences. JESAM, the subject of this paper is another hashed-database
sequence comparison program.
Word-Based Clustering
Word-based clustering is inherently fast because it omits all alignment
steps before clustering. Eliminating initial alignments raises the potential
for poor quality clustering either through poor sensitivity, or poor selectivity.
Word-based clustering uses less information and so has greater error potential,
especially when compared to sequence alignments that can use position-dependent
sequence quality measures. Good specificity depends on refinements such
as statistical weighting schemes where common oligos are given reduced
significance. The sensitivity of word based alignments has not been discussed
to any depth in the literature.
The published work on D-Squared Cluster (Hide et al, 1994, http://www.bchs.uh.edu/stemple/D2/d2.html)
was preliminary and experimental with only limited exploration of the kinds
of optimizations available that might make improvements to the programs
specificity. D-Squared cluster uses windowed (i.e. local) n-mer oligo frequency
comparisons between a query sequence and a compressed database. Using a
Cray Y/MP-48 supercomputer, 884 bases could be compared against 19 million
bases in less than one minute.
RAPID (Rapid analysis of Pre-Indexed Data structures) Miller et al.,
1999 and http://www.bioinf.man.ac.uk/rapid/)
uses oligo frequencies to alter the significance of oligo matches before
scoring particular pairwise matches. No clustering is offered but the program
has been tested on the analogous problem of removing redundancy and contamination
from DNA sequence databases. RAPID uses Receiver Operator Characteristic
(ROC) curves to tune parameters such as oligo size to get database search
results with maximal specificity to the query. Rapid is memory intensive
but relatively fast.
Systems and Methods
The JESAM sequence alignment calculation and storage code is written in
ANSI C with a C++ wrapper for the alignment server. Sequence alignments
were stored in Berkeley DB libraries supplied by Sleepycat: http://www.sleepycat.com/db/.
All the client software is written in JAVA and compiled using Sun Microsystems/Javasoft's
Javac JAVA compilers (http://www.javasoft.com/)
and runs with the Java 1.1 class libraries and Java Virtual Machine JVM
including the implementation in Netscape Communicator 4.5. The IDL interface
specifications were compiled by Object Oriented Concepts' (URL http://www.ooc.com/)
ORBacus IDL to Java and IDL to C++ compilers which have free run-time ORB
licenses. Many ORBs and IDL compilers are available free (URL: http://industry.ebi.ac.uk/~corba/)
as are Sun's Java and IDL compilers. Documentation is distributed throughout
the Java source code in Javadoc comments.
Algorithm
Sequence overlap detection
A multi-stage pipeline was developed to discover all the arrangements of
all pairs of sequences where the alignment could be consistent with the
sequences being cognate and contiguous. The algorithm's first stages reduce
the total number of pairwise sequence alignments whilst aiming to maintain
overlap sensitivity and alignment accuracy.
To ensure that the published alignments were both biologically useful
and mathematically optimal, it was thought necessary to use a dynamic programming
algorithm with a sophisticated gap penalty scheme (Altschul and Erickson,
1986). However, computation time makes it impractical to compare all sequences
against each other for large EST datasets with workstation implementations
of derivatives of the Smith-Waterman algorithm. Specialized hardware was
an unacceptable solution due to perceived problems of cost, availability,
portability, and ease of algorithm development. The JESAM alignment algorithm
therefore uses dynamic programming only for the final alignments relying
on the gross overall overlap being easy to find because the goal was only
to discover potentially overlapping subsequences, not distant homologues
mutated apart though millennia
Alignment Algorithm Stages
Load all non-overlapping 12-mers from all sequences into a hash table
Delete all low-complexity and over-abundant hash table entries
For each sequence
Using a 12-mer window sliding
in single base steps down query
Find the positions of all oligo matches retrieved using 12-mer key
Sort all the oligo matches
by relative start positions (phase) to remove redundancy
Use the representative oligo
matches to seed a windowed optimal alignment
Store all alignments consistent with contiguity
For all the alignments described in this paper, an affine gap scoring
scheme (+5 match, -15 mismatch, -15 gap-start, -15 per-gap-base) was used
with end gaps only penalized if not-consistent with contiguity and an overall
score of 200 needed for an alignment to be stored. To ensure sensitive
alignments are calculated and stored, no predefined repeats are masked.
Furthermore, repeat discovery is easier to do accurately after all alignments
are calculated.
Supercluster creation
A simple clustering algorithm was chosen to summarize groups of overlapping
sequences both as an extension of the alignment database and to prove the
ease of writing client CORBA components. The algorithm aims to create a
series of independent clusters through recursive calls that follow all
alignments for every sequence. Each cluster ultimately contains all the
sequences that can be linked by any series of stored overlaps to every
cluster member. The clustering is performed without regard for any inconsistent
alignment combinations making this algorithm much simpler and less selective
than those used to produce highly optimized clustering databases like UniGene.
These simple superclusters have a bias towards sensitivity and overclumping
that aids further processing (e.g.. reduced likelihood of data loss for
cluster assembly) and directly offer a useful and easily understood benchmark
to contrast against other clustering schemes.
Implementation
JESAM takes novel approaches to both sequence comparison and clustering:
alignments are calculated using a PVM process farm, and published via CORBA.
A Java program acts as a CORBA client to request alignments, and make sequence
clusters and then turns itself into a CORBA server for the cluster data.
Alignment calculation and storage.
All the sequences can be loaded from flat files or from a CORBA sequence
server. The latter would be more convenient but the former offers higher
performance with current sequence server implementations. All sequences
are used to extract non-overlapping 12-mers which are recorded in a hash-table
of linked lists. Abnormally common oligos and low-complexity oligos may
have all their linked lists freed but no predefined repeats are masked.
New sequences are retrieved in batches and overlapping 12-mers are used
to look up oligo hits in the hash table. Oligo hit results are partitioned
between the slaves which do a banded Smith-Waterman-based sequence comparison
looking for matches consistent with sequence overlap. The parallel computer
architecture and all affine gap penalties can be independently adjusted
on the command-line. The slaves receive computational tasks via Parallel
Virtual Machine (PVM) message transfers which are asynchronous and fully
buffered. Buffering is controlled by requiring feedback to the oligo scanner
and limiting the number of outstanding comparisons. To spread the network
load, slaves ask for batches of sequences from a sequence server only when
needed and then cache their set locally. The slaves may need a large amount
of virtual memory, (to cache sequences: one byte per base) but little RAM
(typically 10 to 20 MB) for the actual comparisons because backtracking
memory usage grows linearly with sequence length. The largest memory requirements
are for the oligo hash-table and the Berkeley DB libraries which both have
a random data access pattern: see tables 2 and 3 for actual figures. Cache
sizing is configured on the command line. The minimum cache size is impossible
to estimate without experimentation. A larger cache is needed for alignments
of very repetitive/redundant sequence sets (more matches), slower disk
access and for larger PVM configurations.
The distributed architecture of JESAM provides a flexible framework
to allow extension to more computationally intensive sequence comparisons
(for example base call error probabilities) and could also support other
costly calculations like assembly contig building via directed graph explorations.
The PVM slaves running sequence comparisons are typically CPU-limited so
the performance of the PVM implementation on networks of cheap uniprocessor
PCs is equivalent to that of more expensive multi-processor servers.
Updates do not require recomputation of known alignments because all
sequence overlaps are automatically stored by the Berkeley DB routines.
If a new alignment crosses a user-determined threshold score, then it is
saved complete with the position of all indels, starts and ends. The alignments
are offered by a CORBA interface provided by a C++ wrapper around the same
ANSI C code used to create the alignments.
A Java client recursively steps through the matches to make superclusters
which are stored in a serialized Java Hashtable object which loads and
saves quickly. The Supercluster server took only one afternoon to write
thanks to the alignment data CORBA interface, simple algorithm, and the
innate productivity of Java. Both the alignment and supercluster data can
be viewed interactively using web-accessible Java applets or larger datasets
can be saved to client file systems for use in other file based scripts
etc. The JESAM clusters have been integrated with other databases at the
European Bioinformatics Institute, see URL http://corba.ebi.ac.uk/EST
for browser demonstrations.
Figure 1
Diagram showing how PVM links are used to distribute data and calculations
across a number of processes which may be running on different processors
in different computers.

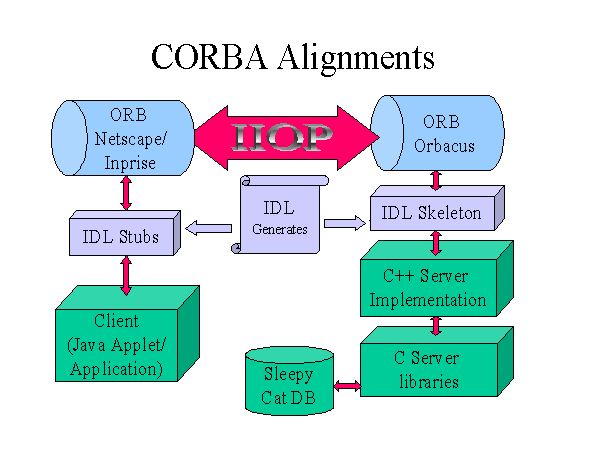
Figure 2
Diagrammatic representation of how an IDL file (centre) is used to create
both the server skeleton code (C++), and the client stub code which in
this case is Java. Other vendor's CORBA products could replace the examples
shown.
Results and Discussion
JESAM Performance
Performing EST comparisons using JESAM can be fast and inexpensive due
to the underlying design: storing results reduces computational needs,
and PVM can spread the incremental calculations across a network of cheap
personal computers, or idle workstations. A total of greater than
120,000 ESTs were aligned and clustered in five hours using a total of
11 processors (UltraSPARC and Compaq Alpha at EBI, all UltraSPARC at Cereon).
Scaling efficiency has been tested in detail (Tables 1,2 and 3) with up
to 40 slaves on a small dataset (120,000 ESTs) but larger datasets allow
greater scaling efficiency (PVM daemon, or network communication restrictions
will appear with less than 100 slaves with many PVM configurations). The
European Bioinformatics Institute can maintain fully updated alignment
and cluster databases (with their Internet accessible interfaces) for four
model organisms (mouse, rat, Aspergillus nidulans, and Zebrafish)
using only six distributed processors (2 in server, 4 in separate slaves)
for one evening every two weeks. These on-line databases are accessible
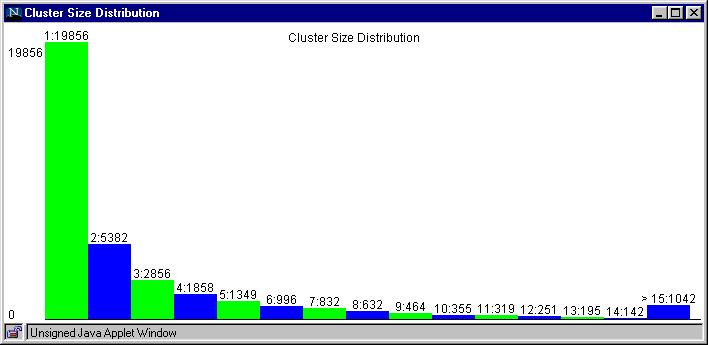
via multiple applets to allow browsing of clustering statistics,
pairwise alignments, cluster details, and downloads of FASTA format
sequences for individual clusters.
Table 1
| Number of Slaves |
10 |
20 |
40 |
| Wall Time (minutes) |
328 |
171 |
107 |
Total time taken to align all 123459 Rat ESTs ( in EMBL and EMBLnew
on 15th September 1999, totaling 48461025 bases) against each other and
then store the alignments when using different numbers of slaves. The master
processes were run on a two processor (400 MHz) UltraSPARC server and the
slaves were run on separate (248 MHz) UltraSPARC servers all running Solaris
2.5 or 2.6.
Table 2
|
Number of Slaves |
| Process |
10 |
20 |
40 |
| Master pvmd3 |
8360 |
29560 |
267280 |
| jesam |
26904 |
26784 |
25016 |
| jesamscanner |
191672 |
192656 |
193288 |
| jesamdistributer |
292912 |
309656 |
455432 |
| Slave pvmd3 |
18224 |
23504 |
61144 |
| jesamslave |
20248 |
17328 |
21896 |
Peak Core Memory Usage (KBytes) for all processes creating the alignments
for Table 1. All processes were sampled at 10-minute intervals. Figures
shown are for the largest memory usage by any instance of each process
at any sample time so actual memory usage is less than shown.
Table 3
|
Number of Slaves |
| Process |
10 |
20 |
40 |
| Master pvmd3 |
9000 |
30200 |
267960 |
| jesam |
27304 |
27184 |
27232 |
| jesamscanner |
192048 |
193032 |
193744 |
| jesamdistributer |
565128 |
588208 |
757264 |
| Slave pvmd3 |
18560 |
24000 |
61640 |
| jesamslave |
20712 |
17576 |
22120 |
Peak Virtual Memory Usage (KBytes) calculated as per Table 2
Clustering Comparisons
A full comparison of all the mentioned EST alignment and clustering programs
on multiple representative datasets is a formidable task and certainly
beyond the scope of this paper. Instead, samples of UniGene Mouse and Rat
databases were studied by comparison to superclusters and alignments produced
by JESAM. The UniGene databases are frequently referenced and important
resources that have guided many large-scale experiments so an investigation
into their contents has potentially significant implications. An initial
investigation suggested it was impossible to use identical datasets for
this comparison because the UniGene databases include sequences which have
been intentionally deleted from the EMBL and GenBank nucleotide databases
and were therefore not publicly retrievable. (A separate ftp site for this
information (ftp.ncbi.nlm.nih.gov/repository/UniGene/ was, however,
identified in review) Unfortunately, the non-identical input sequences
led to frequent differences in the cluster sets and this complicated and
limited the scale of investigations into the basis of the overall cluster
discrepancies. It is worth noting that the more intensive EST/genomic comparisons
performed by Wolfsberg and Landsman were limited (by the inherent complexity
of the underlying data) to analyzing just 15 genes and their study concluded
that this small sample gave indications of uncharacterized alleles, partial
splicing, undocumented splicing, uncoupled clones, and possibly novel biological
phenomena !
The UniGene investigation detailed in Fig. 3 and Fig. 4 was small but
it sampled examples of many of the known or expected properties of the
UniGene databases, and some of the advantages and disadvantages of JESAM's
simplistic superclustering client. UniGene Rat excluded >20,000 sequences
included in the corresponding JESAM alignments and clusters, and UniGene
mouse similarly excluded >150,000 sequences. The two rat databases were
generally more similar than the two mouse databases (data not shown). Perhaps
most significantly, JESAM both clustered more sequences and created more
clusters for both data sets despite the use of genomic clones in UniGene
(excluded from JESAM) that helped clustering in two of the ten mouse clusters
examined (but no rat cluster). However, one rat UniGene cluster (Rn. 34368)
did probably correctly combine two large divergent cDNAs which may encode
alternative transcripts that JESAM rejected as being clearly non-overlapping
sequences. The worst feature of the current published JESAM superclusters
was demonstrated with the 12 sequences in UniGene Mm. 2477 which were combined
into one supercluster of size 116977 with many other sequences presumably
joined on repetitive DNA. This supercluster would not form with a more
selective scoring scheme but sensitivity would then be lost. Biasing towards
sensitivity minimizes information loss yet still significantly reduces
the problem scale for later cluster partitioning software components. With
all the alignments available on-line, it should be relatively easy to develop
and test some quite sophisticated partitioning or assembly algorithms so
these superclusters should not be seen as final answers in contrast to
the other EST cluster databases which cannot be programmatically interrogated.
Figure 3
Comparison between Rat UniGene and EBI Superclusters. First 10 "interesting"
(defined by UniGene) clusters taken from largest (best sampled) Rat cDNA
library: Lib.53, UI-R-C0 containing 7024 sequences. UniGene build 42, uploaded
on Wed Feb 17 1999 (80755 sequences, 23003 clusters including 9520 orphans).
EBI superclusters were made on 19th February 1999 (101383 sequences, 31863
clusters, including 17765 orphans). Comparison done on 22nd February 1999.
NR = Not Retrievable from EMBL, GenBank, or dbEST (i.e. withdrawn entries,
or not available at the time of the study - Nicole Redaschi, personal communication)
|
UniGene ClusterID
|
UniGene
Cluster Size
|
Super
Cluster Size
|
Missed
ESTs
|
Best Missed
|
Explanations
|
|
Rn.12107
|
15
|
17
|
AI228202, AI137581, AA997212, -AA963421 |
AI228202 against AI231460, 124 base overlap with 3 mismatches |
AI228202 was prepended, for unknown reasons, with 5 extra bases "actaa"
on 9th January 1999. These 5 bases reduce the match quality. AA963421 was
NR |
|
Rn.32936
|
10
|
11
|
AA998090 |
AA998090 against X53725, 138 base overlap with two mismatches |
AA998090 contains a low complexity region that may have been masked
in UniGene |
|
Rn.12821
|
7
|
4
|
-AI029388, -AI045806, -AI029890, |
None |
AI029890 was NR so other two form a separate cluster |
|
Rn.11720
|
8
|
13
|
AA899962, AI236745, AI234665, AI170199,
AA945230,
AA943491,
-AI029893 |
AI236745 against AA963979, 75 base overlap with one indel, and one
mismatch |
All found in separate cluster Rn.7578 with extra sequence AI030576
which was NR
AI029893 was NR |
|
Rn.13083
|
6
|
7
|
AB006882 |
AB006882 against AB020504, 510 base overlap with 4 mismatches |
AB020504 has replaced AB006882 |
|
Rn.12740
|
10
|
12
|
AI407336, AI407876 |
AI407336 against AI138051, 384 base overlap with one mismatch |
Both entered GenBank on 9th February, and EMBL on 11th February 1999
but not in dbEST version used (on 13 February according to release notes)
for UniGene Rat. |
|
Rn.11948
|
6
|
9
|
AI406632,
AI408422,
AI409932 |
AI406632 against AA997064, 454 base overlap with one indel and 5 mismatches |
Too recent to appear in this UniGene release |
|
Rn.9416
|
8
|
33
|
Rn.2375 |
AI029424 against U35775, 137 base overlap with one indel and one mismatch |
AI029424 appears to join two alternative 3' ends ? |
|
Rn.8514
|
6
|
7
|
AI407620 |
AI407620 against AI408791, 392 base overlap with one indel and 15 mismatches |
Too recent to appear in this UniGene release |
|
Rn.34368
|
4
|
2
|
-AI043650, -AA964642 |
AA998702 matches AI02983 correctly |
Sequences share a pattern of local similarities inconsistent with contiguity.
UniGene cluster may represent alternative splicing variants. |
Figure 4
Comparison between Mouse UniGene and EBI Superclusters. First 10 "interesting"
(defined by UniGene) clusters taken from largest (best sampled) Mouse cDNA
library: Lib.30, Soares Mouse Embryo NbME13.5 containing 39996 sequences.
UniGene build 46, uploaded on Mon 15th February 1999 (268247 sequences,
15275 clusters including 2761 orphans). EBI superclusters were made on
23rd February 1999 (424080 sequences, 65588 clusters including 42418 orphans).
Comparison done on 24th February 1999. NR = Not Retrievable from EMBL,
GenBank, or dbEST at the time of the study.
| UniGene ClusterID |
UniGene
Cluster Size |
EBI
Cluster Size |
Missed ESTs |
Best Missed |
Explanation
|
|
Mm.2496
|
25
|
30
|
AA048065, AA198448, AA230545, AA967785, W58964, W88178, W98102
-L27220, -AI425958 |
AA967785 against AA218283, 432 base overlap with 2 indels and 29 mismatches |
All 7 missing ESTs were derived from poor quality traces and therefore
excluded from UniGene. L27220 is genomic DNA. AI425958 was NR |
|
Mm.11285
|
14
|
12
|
AA052635, -AI427383, -AI430589, -AI431070 |
AA052635 against AA058152, 140 base overlap with 1 indel and 5 mismatches. |
AA052635 is only 171 bases long and contains a low complexity region.
AI427383, AI430589, and AI431070 were NR |
|
Mm.3501
|
11
|
11
|
AF013117, AA693148, -AI430391, -AI430109 |
AF013117 against D49544, 465 base overlap with 5 mismatches |
AF013117 has been excluded for unknown reasons. AA693148 is annotated
as low quality. AI430391 and AI430109 were NR |
|
Mm.3277
|
12
|
6
|
-AA030514, -AA171281, -AI323427, -AI427499, |
None |
Z22923 is genomic DNA so AA030514, AA171281, AI323427, AI427499 were
split off into separate clusters. |
|
Mm.30219
|
9
|
12
|
AI317572, W78547, W66677, AA237557, AA510343, AI181413, -AI430530,
-AA110526, -AI429692 |
AI317572 against AA049045, 314 base overlap with perfect match |
AI317572, W78547, W66677, AA237557, AA510343, AI181413, were excluded
for unknown reasons. AI430530 andAI429692 were NR. AA110526 is only 84
bases long |
|
Mm.20964
|
11
|
13
|
AA450931, W64306, W64209, AA268149, AA170119, -M36778, -W43961 |
W64306 against W64299, 238 base overlap with 2 indels. |
AA450931, W64306, W64209, AA268149 were excluded for unknown reasons.
M36777 and M36778 are two alternatively spliced mRNAs that differ in over
1KB and are non overlapping. |
|
Mm.3127
|
11
|
16
|
AA271310, AA028577, AA028535, AA062181, AI414081 |
AA271310 against W83829, 263 base overlap with 2 indels, and 8 mismatches |
AA271310, AA028577, AA028535, and AA062181 were excluded for unknown
reasons. AI414081 was entered on 11th February 1999 . |
|
Mm.2477
|
12
|
116977
|
Not relevant |
Not relevant |
Joined on repetitive DNA |
|
Mm.4758
|
9
|
59
|
-X86368, -AI430408, -AI429593 |
AA048965 against L38607, 272 base overlap with 20 mismatches |
Possibly matching on low-copy repetitive DNA. X86368 is genomic DNA,
AI430408 and AI429593 were NR. |
|
Mm.30224
|
7
|
5
|
-AI430492, -AI430463, |
None |
AI430492 and AI430463 were NR |
Figure 5 optional suggestions:
Chooser applet window
Rat Cluster Size Histogram
Extensions
Memory usage could be drastically reduced by switching to a partitioned
sequence database: this would both reduce the number of sequences and oligos
in memory at any one time and also reduce the subset of sorted alignment
results that would need to be cached for random update access. With this
enhancement, JESAM could scale easily to any N2
sequence comparison but also shrink to fit any parallel architecture. If
access to the oligo hash table became limiting with large numbers of processors,
the single oligo hash table lookup function could be rewritten to use multi-threading
but it would then have to run on a multi-processor computer. If greater
speed were needed on very limited hardware, the slave sequence comparisons
could be significantly optimized relative to the current generic implementation,
and multiple oligo hits per pair of sequences (currently just a single
12-mer) could be required before launching a slave comparison (the existing
code currently used to prevent multiple searches on the same diagonal could
be easily extended for this purpose).
The mouse EST and mRNA clusters (downloaded directly from the CORBA
servers) have been successfully assembled using CAP3 by Juha Muilu (personal
communication) as a useful extension of the JESAM superclusters. CAP3 however
ignores the pairwise alignment data so an opportunity still exists for
a more integrated, higher performance assembly tool tailored to EST data.
A suitable post-assembly goal would be something like the Pangea CRAW tools
(Burke et al., 1998 and Chou and Burke, 1999) which demonstrate how complicated
patterns of expression variations can be summarized and presented in a
Java applet alongside supporting analyses.
Linking the EST clusters with other databases would make them more valuable.
The EMBL outstation European Bioinformatics Institute (EBI) amongst others
(including the OMG's Life Science Research (LSR) Domain Task Force (DTF)
(URL: http://lsr.ebi.ac.uk/) are working towards standards for object interfaces
and components for many kinds of biological data. Some of the EBI's effort's
include physical and genetic maps (Jungfer and Rodriguez-Tomé, 1998),
annotated DNA and protein sequences, and sequence traces (Parsons et al.,
1999).
JAVA RMI could have offered a CORBA alternative but was not investigated
due to the lack of relevant biological standards efforts, frameworks, language
independence, services and local support. AcePerl (Stein and Mieg, 1998),
is another potential alternative.
Conclusion
As explained in the introduction, no EST clustering database can be expected
to provide a perfect view of an organism's genes but the imperfect result
is still valuable. The human UniGene database has been a notably effective
resource for many large and varied scientific studies however this small
study shows the mouse and rat UniGene clusters have questionable features:
the databases sometimes contain deleted sequences, and conversely may exclude
apparently unique and high-quality sequences, and many with perfect matches
to those included sequences. The simple JESAM superclusters have problems
of their own and consequently complement rather than supersede the UniGene
databases but this is a significant role. Furthermore, the public CORBA
interface to model organism alignment data offers great potential for future
enhancements through new public software components interacting over the
Internet, or (since all the code is available and portable) combining public
and private data behind corporate firewalls.
The use of CORBA interfaces in the JESAM package requires more
software libraries (and greater expertise to install and maintain them)
than would a normal file-based Input/Output system but they also offer
greater flexibility. Furthermore, by maintaining all information accessible
online, it is possible to give users an evidence trail that they can follow
all the way from cluster, to alignment, to sequence and even back to the
original sequence trace.
There are currently a few problems deploying CORBA based applications
over the Internet. These problems include: firewalls blocking the IIOP
protocol, the need to download ORB classes to clients due to the lack of
a guaranteed local ORB, and a lack of support for multiple applet signing
which would allow applets to follow object references to objects on computers
other than the original applet's host. Some of the Java and CORBA problems
should decrease once JAVA 2 from Javasoft (URL:http://www.javasoft.com/)
becomes a standard component of every client workstation. Amongst the many
improvements over Java 1.1 is a built-in CORBA ORB allowing distributed
computing to extend out of the Web browser and directly into a user's general
molecular biology application set.
References
Aaronson,J.S., Eckman,B, Blevins,R.A., Borkowski,J.A.,Myerson,J.,Imran,S.
and Elliston,K.O. (1996) Toward the development of a gene index to the
human genome: an assessment of the nature of high-throughput EST sequence
data. Genome Res. 6, 829-845.
Adams et al., (1995), Initial assessment of human gene diversity and
expression patterns based upon 83 million nucleotides of cDNA sequence,
Nature, 377: Supplement 3-174
Altschul, S.F., and Erickson, B.W., 1986, Optimal Sequence Alignments
using Affine Gap Costs, Bulletin of Mathematical Biology, 48, 603-616.
Altschul S.F., Madden T.L. Schaffer, A.A., Zhang, J., Zhang, Z., Miller,
W., and Lipman, D.J., 1997, Gapped BLAST and PSI-BLAST: a new generation
of protein database search programs, Nucleic Acids Res., 25, 3389-3402.
Bailey, L.C, Searls, D.B, and Overton, G.C., 1998, Analysis of EST-Driven
Gene Annotation in Human Genomic Sequence, Genome Res., 8, 362-376.
Battey, J., Jordan, E., Cox, D., and Dove, W., 1999, An action plan
for mouse genomics, Nature Genetics, 21, 73-75
Banfi, S., Borsani, G., Rossi, E., Bernard, L., Guffanti, A., Rubboli,
F., Marchitiello, A., Giglio, S., Coluccia, E., Zollo, M., Zuffardi, O.,
and Ballabio, A., 1996, Identification and mapping of human cDNAs homologous
to Drosophila mutant genes through EST database searching. Nature Genetics,
13, 167-174
Benson, D.A., Boguski, M.S., Lipman, D.J., Ostell, J., and Ouellette,
B.F.F., 1998, GenBank, Nucleic Acids Res., 26: 1-7
Bishop, M., and Thompson, E., 1984, Fast Computer Search for Similar
DNA Sequences, Nucleic Acids Res., 12, 5471-5474
Blake, J.A., Eppig, J.T., Richardson, J.E., Davisson, M.T. and the
Mouse Genome Informatics Group, 1998, The Mouse Genome Database (MGD):
a community resource, Status and enhancements., Nucleic Acids Res., 26,
130-137
Boguski, M., Lowe, T.M.J, and Tolstoshev, C.M., 1993, dbEST - database
for "Expressed Sequence Tags", Nature Genetics, 4, 332
Boguski, M. and Schuler G., (1995). ESTablishing a human transcript
map. Nature Genetics, 10, 369-371
Bonaldo, M.F, Lennon G., and Soares, M.B., (1996), Normalization and
Subtraction: Two Approaches to facilitate Gene Discovery, Genome Res.,
8, 791-806
Bonfield, J.K., Smith, K.F., and Staden R., 1995, A new DNA sequence
assembly program. Nucleic Acids Res., 24: 4992-4999
Burke, J., Wang, H., Hide, W., and. Davison, D.B., (1998), Alternative
Gene Form Discovery and Candidate Gene Selection from Gene Indexing Projects,
Genome Res. 8, 276-290.
Davidson, D., Bard, J., Brune, R., Burger, A., Dubreuil, C., Hill,
W., Kaufman, M., Quinn, J., and Baldock, R., 1997, The mouse atlas and
graphical gene-expression database, Cell and Developmental Biology, 8,
509-517.
Chou, A. and Burke J., 1999, CRAWview: for viewing splicing variation,
gene families, and
polymorphism in clusters of ESTs and full-length sequences. Bioinformatics,
15, 376-381
Eckman, BA, Aaronson, JS, Borkowski, JA, Bailey, WJ, Elliston, KO,
Williamson, AR, Blevins,
RA, (1998), The Merck Gene Index browser: an extensible data integration
system for gene finding,
gene characterization and EST data mining, Bioinformatics, 14: 2-13
Ewing, B. and Green, P., 1998, Base-Calling of Automated Sequencer
Traces Using Phred. II. Error Probabilities. Genome Res., 8:
186-194
Gautheret, D., Poirot, O., Lopez, F., Audic, S., and Claverie, J.,
(1998), Alternate Polyadenylation in Human mRNAs: A Large-Scale Analysis
by EST Clustering, Genome Res., 8: 524-530.
Gerhold, D. and Caskey, T., 1996, It's the genes! EST access to human
genome content, BioEssays, 18, 973-981
Gill, RW, Hodgman, TC, Littler, CB, Oxer, MD, Montgomery, DS, Taylor,
S, Sanseau, P (1997), A new dynamic tool to perform assembly of expressed
sequence tags (ESTs) Comput. Applic. Biosciences 13: 453-457
Graul RC, Sadée W., (1997), Evolutionary relationships among
proteins probed by an iterative neighborhood cluster analysis (INCA). Alignment
of bacteriorhodopsins with the yeast sequence YRO2. Pharmaceutical Research,
14: 1533-1541
G. Grillo, M. Attimonelli, S. Liuni and G. Pesole, (1996), Cleanup:
a fast computer program for removing redundancies from nucleotide sequence
datatbases, Comput. Applic. Biosci., 12, 1-8
Hardison, R.C., Oeltjen, J., and Miller W., 1997, Long Human-Mouse
Sequence Alignments Reveal Novel Regulatory Elements: A Reason to Sequence
the Mouse Genome, Genome Res., 7, 959-966
Hillier L., Lennon G. Becker, M., Bonaldo, M.F., Chiapelli, B., Chissoe,
S., Dietrich, N., DuBuque, T., Favello, A., Gish, W., Hawkins, M., Hultman,
M., Kucaba, T., Lacy,M., Le, M., Le, N., Mardis, E., Moore, B., Morris,
M., Parsons, J., Prange, C., Rifkin, L., Rohlfing, T., Schellenberg, K.,
Bento Soares, B., Tan, F., Thierry-Mieg, J., Trevaskis, E., Underwood,
K., Wohldman, P., Waterston, R., Wilson, R., Marra, M. (1996), Generation
and Analysis of 280,000 Human Expressed Sequence Tags. Genome Res.,
6,
807-828
Hide W, Burke J, Davison DB, (1994), Biological evaluation of d2, an
algorithm for high-performance sequence comparison, J. Comput Biol 1: 199-215
Houlgatte, R., Mariage-Samson, R., Duprat, S., Tessier, A., Bentolila,
S., Lamy, B., and Auffray, C., (1995), The Genexpress Index: A Resource
for Gene Discovery and the Genic Map of the Human Genome, Genome Res.,
5, 272-304
Hunkapiller, T., Kaiser, R.J., Koop, B.F., and Hood, L., 1991, Large-Scale
and Automated DNA Sequence Determination, Science, 254, 59-67
Lijnzaad, P., Helgesen, C., and Rodriguez-Tomé, P., 1998, The
Radiation Hybrid Database, Nucleic Acids Res., 26, 102-105
Hu,J., Mungall, C., Nicholson,D., and Archibald,A.L., 1998, Design
and implementation of a CORBA-based genome mapping system prototype, Bioinformatics,
14,
112-120.
Huang, X., 1996, An Improved Sequence Assembly Program, Genomics, 33,
21-31.
Jungfer, K., and Rodriguez-Tomé, P., 1998, Mapplet: a CORBA-based
genome map viewer, Bioinformatics, 14, 734-738
McCarthy, L.C., Terret, J., Davis, M.E., Knights, C.J., Smith, A.L.,
Critcher, R., Schmitt, K., Hudson, J., Spurr, N.K., and Goodfellow P.N.,
1997, A first-Generation Whole Genome-Radiation Hybrid Map Spanning the
Mouse Genome, Genome Res., 7, 1153-1161
Makalowski, W., Zhang, J., and Boguski, M.S., 1996, Comparative Analysis
of 1196 Orthologous Mouse and Human Full-length mRNA and Protein Sequences,
Genome Res., 6, 846-857
Jiang, J, and Jacob, H.J., 1998, EbEST: An Automated Tool Using Expressed
Sequence Tags to Delineate Gene Structure, Genome Res., 8, 268-275
Küster, B. and Mann, M., 1998, Identifying proteins and post-translational
modifications by mass sprectrometry, Current Opinion in Structural Biol.,
8, 393-400
Lopez, A.J., 1998, Alternative Splicing of pre-mRNA: Developmental
Consequences and Mechanisms of Regulation, Annu. Rev. Genetic., 32, 279-305
Marra, M., Hillier, L., Kucaba, T., Allen,M., Barstead, R., Beck, C.,
Blistain, A., Bonaldo, M., Bowers, Y., Bowles, L., Cardenas, M., Chamberlain,
A., Chappell, J., Clifton, S., Favello, A., Geisel, S., Gibbons, M., Harvey,
N., Hill, F., Jackson, Y., Kohn, S., Lennon, G., Mardis, E., Martin, J.,
Mila, L., McCann, R., Morales, R., Pape, D., Person, B., Prange C., Ritter.,
E., Soares, M., Schurk, R., Shin, T., Steptoe, M., Swaller, T., Theising,
B., Underwood, K., Wylie, T., Yount, T., Wilson, R., Waterston, R., 1999,
An encyclopedia of mouse genes, Nature Genetics, 21, 191-199
Miller, C., Gurd, J and Brass, A, 1999, A RAPID algorithm for sequence
database comparisons: application to the identification of vector contamination
in the EMBL databases, Bioinformatics, 15, 111-121
Miller, M.J., and Powell, J.I., (1994), A Quantitative Comparison of
DNA Sequence Assembly Programs, J. Comput. Biol., 4, 257-269
Myers, Eugene W., (1995) Toward Simplifying and Accurately Formulating
Fragment Assembly, Journal of Computational Biology 2: 275-290.
Orfali,R., and Harkey,D. (1997) Client/Server Programming with JAVA
and CORBA. John Wiley & Sons, New York, N.Y. 10158-0012.
Oliver, S.G., 1996, From DNA sequence to biological function, Nature,
379, 597-600
Orfali,R., and Harkey,D. (1998) Client/Server Programming with JAVA
and CORBA. 2nd Edition, John Wiley & Sons, New York, N.Y. 10158-0012.
Parsons, J.D., (1995) Improved Tools for DNA Comparison and Clustering,
Comput.
Applic. Biosci., 11, 603-613
Parsons, J.D., Buehler, E., and Hillier, L., (1999) DNA Sequence Chromatogram
Browsing Using JAVA and CORBA., Genome Res., 9, 277-281.
Parsons, J.D., Brenner, S. and Bishop, M.J., (1992), Clustering cDNA
Sequences, Comput. Applic. Biosci., 8, 461-466
Pearson, W. R., Wood, T., Zhang, Z., and Miller, W. (1997) Comparison
of DNA Sequences with Protein Sequences, Genomics 46, 24-36
Rounsley_SD, Glodek_A, Sutton_G, Adams_MD, Somerville_CR, Venter_JC,
Kerlavage_AR, (1996), The construction of Arabidopsis expressed sequence
tag assemblies - A new resource to facilitate gene identification, Plant
Pysiology, 112, 1177-1183
Schuler et al. 1996, A Gene Map of the Human Genome, Science, 274,
540-546
Stein L.D. and Thierry-Mieg, 1998, Scriptable Access to the Caenorhabditis
elegans Genome Sequence and Other ACEDB Databases, Genome Res., 8:1308-1315
Stoesser, G., Moseley, M.A., Sleep, J., McGowran, M., Gracia-Pastor,
M., and Sterk, P. (1998) The EMBL Nucleotide Sequence Database, Nucleic
Acids Res., 26, 8-15
Sutton G., White O., Adams M. and Kerlavage A. (1995). TIGR Assembler:
A new tool for assembling large shotgun sequencing projects. Genome Science
& Technology, 1, 9-19.
Wolfsberg, T.G. and Landsman, D., 1997, A comparison of expressed sequence
tags (ESTs) to human genomic sequences, Nucleic Acids Res., 25, 1626-1632
Yee, D.P., and Conklin, D., (1998), Automated clustering and assembly
of large EST collections, proceedings ISMB-98
Acknowledgements
The authors are grateful to Tom Flores for helping to start the CORBA element
of this project. The authors would also like to thank David Starks-Browning
at the EBI, and the Systems staff at Cereon for their support. This work
was funded by EU grant BIO 4 CT 960346